Worksharing: questions and answers
HumanRegulatory and procedural guidance
This page lists questions that marketing-authorisation holders (MAHs) may have on worksharing. It provides an overview of the European Medicines Agency's position on issues that are typically addressed in discussions or meetings with MAHs in the post-authorisation phase. Revised topics are marked 'New' or 'Rev.' upon publication.
A PDF version of the entire post-authorisation guidance is available:
Updated 25/03/2025:
Questions marked with "Rev. Mar 2025"
These questions and answers have been produced for guidance only and should be read in conjunction with the rules governing medicinal products in the European Union, volume 2, notice to applicants.
MAHs must in all cases comply with the requirements of Community legislation. Provisions that extend to Iceland, Liechtenstein and Norway by virtue of the European Economic Area agreement are outlined in the relevant sections of the text.
Mandatory worksharing
In accordance with Article 20(1) of the Commission Regulation (EC) N° 1234/2008, as amended (the ‘Variations Regulation’), a Marketing Authorisation Holder (MAH) shall follow the worksharing procedure and submit the same Type IB or Type II variation, or the same group of variations affecting more than one marketing authorisation from the same MAH in one application.
Applicants belonging to the same mother company or group of companies and applicants having concluded agreements or exercising concerted practices concerning the placing on the market of the medicinal product(s) concerned, have to be taken as “the same marketing authorisation holder”.
Extensions of Marketing Authorisations are excluded from worksharing.
Based on Articles 7, 7a and 20 of the Variations Regulation, when a group of variations only consists of
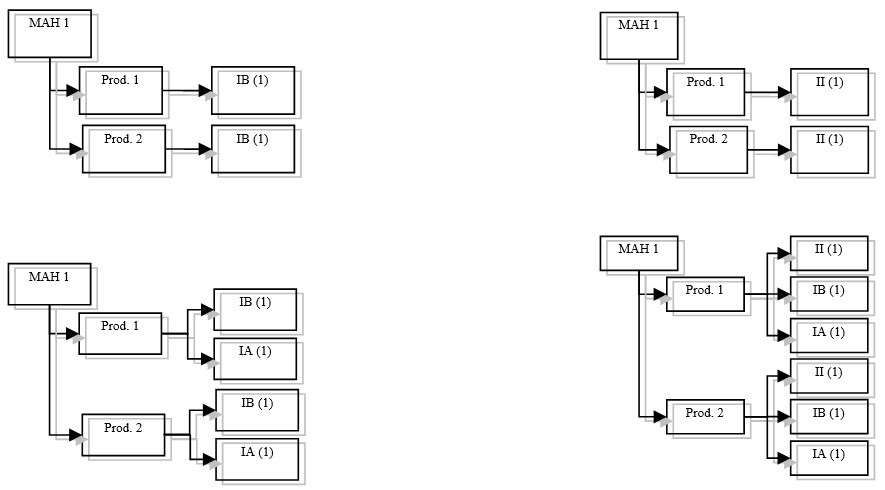
A worksharing procedure can include centralised authorised products (CAP), decentralised procedure/mutual recognition products or purely national marketing authorisations.
In order to avoid duplication of work in the evaluation of such variations, a worksharing procedure has been established under which one authority acts as the ‘reference authority’and examines the variation(s) on behalf of the other authorities. Where at least one of the marketing authorisations concerned is a CAP, the Agency will act as reference authority. The procedure to choose the reference authority where the worksharing procedure does not involve a CAP is described in CMDh BPG on Worksharing (Chapter 7).
Voluntary worksharing
Additionally, in accordance with Article 20(11) of the Commission Regulation (EC) N° 1234/2008), as amended, in justified cases agreed by the Agency (and competent authorities of the Member States, if applicable) holders may choose to follow the worksharing procedure also where a minor variation of type IB or a major variation of type II, or a group of variations where at least one of the variations is a minor variation of type IB or a major variation of type II, that does not contain any extension, relates to several marketing authorisations owned by several holders.
References
In a worksharing procedure, it is required that the same change(s) will apply to the different medicinal products concerned from the same MAH, with either no or limited need for assessment of a potential product-specific impact. Therefore, where the ‘same’ change(s) to different marketing authorisations require the submission of individual supportive data sets for each medicinal product concerned which each require a separate product-specific assessment, such changes will not fall under the mandatory worksharing.
Grouped variations should be subject to a worksharing procedure, provided that the same group of variations applies to all medicinal products concerned by the worksharing procedure.
Non-exhaustive examples of changes which should be submitted for evaluation under worksharing (if appropriate) are listed below:
Clinical/Pharmacovigilance
Quality
In order to facilitate the planning of a worksharing procedure, MAHs are advised to inform the Agency at least 2 months in advance of the submission of a variation/group of variations to be subject to a worksharing procedure, by means of a ‘letter of intent’.
The ‘letter of intent’ should provide the following information:
A ‘letter of intent template’ is available on the Agency’s website. To submit your request, raise a ticket via EMA Service Desk. Please click on “Finance Services”, then the Type of ticket request to be selected is “Request for high-level procedure or ASMF number” followed by sub-option “Workshare Procedure number”. The letter of intent should be attached to the EMA Service Desk ticket.
If you do not have an EMA Account, you may create one via the EMA Account Management portal.
Upon receipt of the letter of intent, the Agency will review and decide whether the proposed worksharing procedure is acceptable. Subsequently, the Agency will initiate the Rapporteur appointment procedure.
Following an ‘Expression of Interest’ a Rapporteur (and, if relevant, a Co-Rapporteur when the application includes a new indication) will be appointed for the procedure. It is expected that the (Co-)Rapporteur will be one of the Rapporteurs of the centrally authorised medicinal products or a CHMP member representing one of the RMSs or National Competent Authorities for the nationally authorised products. The MAH will be informed accordingly.
A shorter pre-submission phase is envisaged, in cases where:
Worksharing procedure for multiple centrally authorised medicinal products (‘duplicates’)
The submission of a formal letter of intent is not required, however applicants are advised to request a WS number. The request should be submitted by raising a ticket via EMA Service Desk. Please click on “Finance Services”, then the Type of ticket request to be selected is “Request for high-level procedure or ASMF number” followed by sub-option “Workshare Procedure number” detailing the list of products, the intended submission date and the scope of variation they are planning to apply for (a draft cover letter is also accepted). Marketing Authorisation Holders are advised to submit such variations as usual.
The submission requirements as set-out in the PAG sections for the different types of variations will also apply to variations subject to worksharing, but the application should be provided as one integrated submission package (eCTD sequence) per product, covering all variations applied for. Please refer to the eCTD Variations Q&A document, for guidance on the submission of variations in eCTD format.
This will include a cover letter and electronic application form, together with separate supportive documentation for each medicinal product concerned and revised product information/risk management plan (if applicable) for each medicinal product concerned.
For queries relating to the presentation of the application, please contact the Agency (Contacting EMA: post-authorisation).
References
The worksharing application must be submitted at the same time to all relevant authorities, i.e. in case the application consists of centrally and nationally authorised medicinal products, the submission should be made to EMA only using the eSubmission Gateway. All NAP submissions (worksharing containing at least 1 CAP) sent to EMA via eSubmission Gateway/Web Client will be considered delivered to all Competent Authorities representatives, alternates and experts of the scientific committees. All EMA submissions should be sent via EMA eSubmission Gateway/Web Client only.
Submission to the European Medicines Agency
The use of the eSubmission Gateway or Web Client is mandatory for all electronic Common Technical Document (eCTD) submissions through the centralised procedure. This applies to all applications for human medicines.
More information on how to register and connect to the Gateway / Web Client can be found in the eSubmission website and detailed information on how to submit can be found in eSubmission Gateway guidance documents.
An automated acknowledgement is sent from the system confirming whether the submission has passed the relevant technical validation criteria and whether it has been uploaded to the Agency’s review tool and made available via the Common Repository.
Where applicable, revised product information Annexes (including Annex A, if applicable) should be included in electronic (Word and PDF) format in the same eSubmission Gateway or eSubmission Web Client package within a folder called ‘working documents’. Where applicable changes in Word documents should be indicated using ‘Tools-Track Changes’. Clean PDF versions should have all changes ‘accepted’.
For Centrally Authorised medicinal products (eCTD mandatory)
An electronic copy containing the relevant eCTD sequence for each product, should be submitted to the Agency. The coordinating Product Lead (if worksharing procedure contains at least one Type II variation) or else the appointed Procedure Manager should be indicated in copy (“cc”) on the cover letter.
For nationally authorised medicinal products (eCTD)
eSubmission Gateway / Web Client package of the Variation application form and supportive documentation for each product should be submitted to the Agency in accordance with the “Dossier Requirements for referral, ASMF and NAP submissions (PASS107, Workshare, Signal Detection procedures) and ancillary medicinal substances in a medical device” document. Paper submissions are not accepted.
Submission to the Competent Authorities of the Member States
Where nationally authorised medicinal products are part of the worksharing, the applications are submitted to the Agency only via the eSubmission Gateway and there should not be additional parallel submissions to Member States, even if some products are not relevant to some MSs. All submissions are available to all Competent Authorities of the Member States via the Common Repository. The Common Repository provides access to all involved Parties (the Agency, Member States and Committee Members) to receive the full data for the worksharing application.
If amendments are requested by the Agency as a result of the validation, updated documentation should also be submitted via the eSubmission Gateway/Web Client and it will be available to the network via the Common Repository.
Submission to the Rapporteur and Committee members
All submissions sent to EMA via eSubmission Gateway/Web Client will be considered delivered to all Competent Authorities’ representatives and alternates.
The dossier requirements for post-authorisation submissions in the centralised procedure should be followed.
For a full overview of dossier requirements for Competent Authorities of Member States (Co-)Rapporteur and Committee members, including delivery addresses, please refer to the following document: Dossier requirements for Centrally Authorised Products (CAPs).
For requirements for non-eCTD format submissions, please refer to the “Dossier requirements for nationally authorised products (referral, PASS107, workshare, signal detection procedures) and ancillary medicinal substances in a medical device” document.
References
The Agency will allocate a 'high-level' cross-products procedure number, which will be used for the handling of worksharing procedures affecting more than one medicinal product. A new procedure code (abbreviation) is used for worksharing procedures i.e. “WS”. As the 'high-level' number cannot be allocated to one single product, the procedure number will therefore contain “xxxx” as a place-holder for the product number.
Example: EMEA/H/C/xxxx/WS/0003
For each medicinal product concerned by the worksharing procedure, the following worksharing number (which includes a reference to the “WS” procedure to which it belongs) will be allocated:
Example: EMEA/H/C/prod_nb/WS0003/nn which was submitted as part of the 3rd worksharing procedure received by the Agency “WS0003”
Worksharing applications for a group of variations will include the suffix “/G” e.g. EMEA/H/C/ xxxx/WS/0004/G and EMEA/H/C/prod_nb/WS0004/nn/G.
For all worksharing procedures, including those which contain nationally authorised medicinal products, the ‘high-level’ procedure number should be systematically obtained from the Agency shortly before submission by sending your request via EMA Service Desk with a letter of intent, see question “What pre-submission steps will apply to a worksharing procedure?”.
The MAH must submit the variation application for worksharing, at the latest by the recommended submission dates published on the Agency’s website (See also Human Medicines – Procedural Timetables / Submission dates).
As a general rule, worksharing procedures will follow the assessment period of the highest type of variation included.
If a submission for a worksharing application does not include all marketing authorisations owned by the same holder affected by the proposed changes, the holder will have to revise its application to include all affected marketing authorisations.
In general, variations submitted for worksharing will follow the 60-day evaluation timetable of Type II variations and weekly-start timetables may apply to the assessment following the same principles as those applied to the assessment of Type II variations. The 60-day period may be reduced having regard to the urgency of the matter, particularly for safety issues, or may be extended to 90 days for Type II variations concerning changes or additions to the therapeutic indication or for grouping of variations accepted by the agency and not listed in Annex III of the Variations Regulation.
Type IB worksharing procedures with type IB as the highest type of change applied will follow a 30-day timetable. However, in specific cases this can be extended to a 60 or 90-day timetable if needed.
For the detailed evaluation timetable, of Type IB and Type II Worksharing procedures, please refer to the PAG for Type II variations “How shall my Type II application be handled (timetable)?”
Upon finalisation of the review of the variations subject to the worksharing procedure, the Agency will issue an opinion reflecting the final outcome of the procedure. Such opinion will also list any variations (e.g. as part of a group, or for a specific medicinal product) which are not considered approvable, unless they had been withdrawn by the holder during the procedure. The same general principles as for grouped variations apply - see the PAG on grouping “What will be the outcome of the evaluation of a grouped variation application”?
Schematic structure of the CHMP Opinion and Annexes for an application under worksharing, consisting of centrally and nationally authorised medicinal products:
Note:
Upon adoption of the CHMP Opinion on the worksharing procedure, the Agency will inform the MAH and Member States concerned (if applicable) as to whether the opinion is favourable or unfavourable (including the grounds for the unfavourable outcome), as well as whether the Commission Decision granting the Union marketing authorisations require any amendments.
Where the outcome of the procedure is favourable and the Commission Decision granting the Marketing Authorisation requires amendments, the Agency will inform the Commission accordingly.
Re-examination
Article 9(2) of Regulation (EC) No 726/2004, also applies to CHMP Opinions adopted for worksharing procedures. This means that the MAH may give written notice to the Agency/CHMP that he wishes to request a re-examination within 15 days of receipt of the opinion (after which, if he does not appeal, the opinion shall be considered as final). The grounds for the re-examination request must be forwarded to the Agency within 60 days of receipt of the opinion. In case the MAH requests that the committee consults a SAG in connection with the re-examination, the applicant should inform the CHMP as soon as possible of this request.
The CHMP will appoint a different (Co-) Rapporteur, to co-ordinate the re-examination procedure. Within 60 days from the receipt of the grounds for re-examination, the CHMP will consider whether its opinion is to be revised. If considered necessary, an oral explanation can be held within this 60-day timeframe.
EMA charges a fee for a re-examination of an opinion. For more information, please refer to the Fee Q&As in Annex IV, Section 4, on Fees payable to the European Medicines Agency.
Decision-Making Process for centrally authorised medicinal products
Upon receipt of a favourable CHMP opinion which requires amendments to the decision granting the marketing authorisation, the Commission shall amend the marketing authorisation for each centrally authorised medicinal product to reflect the approved variation(s) within 2 months, for the variations listed under Article 23(1a)(a) or within one year for the other variations. A single decision will be issued for each centrally authorised medicinal product.
Article 23(1a)(a) provides for a two month timeframe for amending the decision granting the marketing authorisation for the following variations:
All the other variations will follow a yearly timeframe for update of the respective Commission decision.
The Agency applies the existing post-opinion timeframes, as set-out in The linguistic review process of product information in the centralised procedure – human. The QRD linguistic check will be performed on one set of Annexes of one centrally authorised medicinal product. In case of comments, it will be up to the MAH to correctly implement the same amendments in the other centrally authorised products, as appropriate.
The Agency, in cooperation with the QRD members and the MAH will aim at providing final, checked translations for all centrally authorised products included in the worksharing procedure to the MAH at opinion stage in case of a worksharing procedure for a Type IB variation or by Day +27 in case of a worksharing procedure for a Type II variation. (See also: “When do I have to submit revised product information? In all languages?”).

MA updating Process for nationally authorised medicinal products (if applicable)
Upon receipt of the final opinion, the Member States concerned shall approve the final opinion, inform the Agency accordingly and where necessary, amend the national marketing authorisations within 60 days, provided that the documents necessary for the amendment of the marketing authorisation have been transmitted to the Member States concerned.
Implementation
Type IB variations approved via a worksharing procedure, may be implemented upon receipt of the favourable CHMP opinion.
Variations listed in Article 23(1a)(a) may only be implemented once the Commission has amended the marketing authorisation and has notified the MAH accordingly.
Type II variations approved via a worksharing procedure, which do not require any amendment of the marketing authorisation or which follow a yearly update of the respective Commission Decision can be implemented 30 days after receipt of the favourable CHMP opinion.
The agreed change(s) should be included in the Annexes of any subsequent regulatory procedure.
Variations related to safety issues, including urgent safety restrictions, must be implemented without delay, within a timeframe agreed by the marketing authorisation holder and the Agency.
References
For information on fees to be paid, applicable fee reductions and payment process, please refer to the Fee Q&As in Annex I, Section 5, on the Fees payable to the European Medicines Agency.
There is no fee payable for Type IB worksharing applications.
References
If the variations subject to worksharing affects the summary of product characteristics (SmPC), labelling or package leaflet, the revised product information annexes must be submitted as follows:
a. Worksharing procedure for type-II variations
At submission (day 0):
After CHMP opinion (day +5):
After linguistic check (day +25):
Only one centrally authorised medicinal product will undergo a linguistic check. In the cases where the changes to the product information may vary between products, the product with the most complex changes will generally be the one subject to linguistic check.
b. Worksharing procedures for type-IB variations
At submission (day 0):
Day +25 after start of procedure:
For such procedures, a linguistic review will take place in parallel to the scientific assessment. It is therefore expected that the texts provided at day +25 after start of procedure will be the final texts.
Overview
| Day | Languages* | Type-II variation(s) | Type-IB variation(s) |
|---|---|---|---|
| 0 | English | Electronically Word format (highlighted) All CAPs | Electronically Word format (highlighted) All CAPs |
| Other European Economic Area | - | Electronically Word format (highlighted) One CAP | |
| +5 | All European Economic Area | After opinion Electronically Word format (highlighted) One CAP | / |
| +25 | All European Economic Area | After opinion Electronically Word format (highlighted) PDF format (clean) All CAPs | After start of procedure Electronically Word format (highlighted) PDF format (clean) All CAPs |
*Complete set of annexes i.e. annexes I, II, IIIA and IIIB submitted as one document per language.
The 'complete set of annexes' includes annexes I, II, IIIA and IIIB, i.e. all SmPC, labelling and package-leaflet texts for all strengths and pharmaceutical forms of the product concerned, as well as annex II.
The complete set of annexes must be presented sequentially (i.e. annex I, II, IIIA, IIIB) as one document for each official EU language. Page numbering should start with '1' (bottom, centre on the title page of annex I). The Quality Review of Documents (QRD) convention to be followed for the European Medicines Agency QRD templates should be followed. When submitting the full set of annexes in PDF format, this should be accompanied by the completed Formatting check list for the product information (annexes) in PDF format - veterinary and following the User guide on how to generate PDF versions of the product information - human.
The electronic copy of all languages should be provided as part of the variation application in the eCTD for the product concerned, on Gateway / Web Client . Highlighted changes should be indicated via 'Tools – Track changes'. Clean versions should have all changes 'accepted'.
Icelandic and Norwegian language versions must always be included.
The annexes provided should only reflect the changes introduced by the variation concerned. However, in exceptional cases where MAHs take the opportunity to introduce minor linguistic amendments in the texts (e.g. further to a specimen check) this should be clearly mentioned in the cover letter and in the scope section of the application form.
In addition, the section 'present / proposed' in the application form should clearly list the minor linguistic amendments introduced for each language. Alternatively, such a listing may be provided as a separate document attached to the application form. Any changes not listed will not be considered as part of the variation application.
In such cases, and in cases where any other ongoing procedures may affect the product information annexes, the MAH is advised to contact the Agency in advance of submission or finalisation of the procedure concerned.
For variations that affect annex A (e.g. introduction of a new presentation), the following principles apply:
Upon adoption of the opinion, the Agency will prepare and send the revised English annex A to the MAH for each CAP reflecting the new or amended presentation.
After CHMP opinion (day +5), the MAH will provide the Agency with the electronic versions of the complete set of annexes in all languages as well as the translations of the revised annex A for each CAP as a separate Word document.
Please be reminded that in accordance with Union data protection requirements, no personal data should be included in the annotated PIs. This applies to the English version submitted at the time of opinion, the draft translation versions of the PI in all languages submitted at D+5 as well as the final translations submitted at D+25. Please submit annotated PIs in an anonymised format (i.e. names of the reviewers removed from the track-changes). The annotated product information files must include the statement containing the procedure number(s) and may be published on the EMA website as part of the product EPAR page. If you do not wish to do so, please ensure that the individuals whose data is included consented to its sharing with EMA, the publication on the EMA website and its further sharing by EMA with third parties such as other marketing authorisation applicants, marketing authorisation holders and National Competent Authorities, as relevant. EMA expressly disclaims any liability or accountability for the presence of unnecessary personal data in the annotated PI submitted by the marketing authorisation holder.
References